
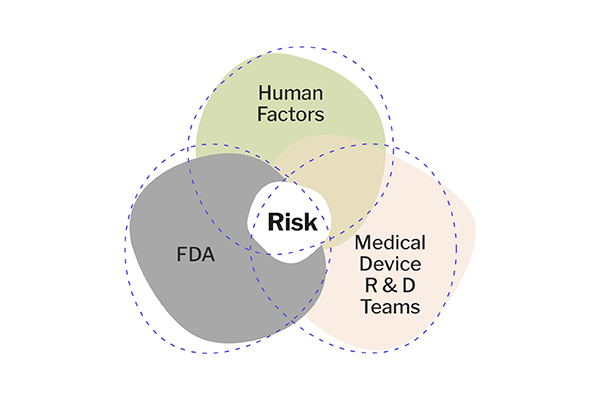
Human Factors, Medical Device R&D Teams, and the FDA: How Do We All Get on the Same Page About Risk?
Medical device manufacturers face challenges daily when submitting medical care devices to the Food and Drug Administration (FDA) for approval and maintaining the quality and safety of an existing device.
The difficulty in medical device R&D stems from the fact that we are trying to develop complex devices in a highly regulated field, requiring large, multi-disciplinary product development teams with competing priorities. Communication barriers between regulatory, clinical, quality, engineering, marketing, and business teams occur regularly, causing common goals to get lost amidst varying priorities.

Listening to FDA presentations and conversations among medical device manufacturing teams over the years has brought to light three key points:
· The FDA continues to increase its focus on continuous risk and quality assessments.
· There are different perspectives, nomenclature, and priorities across medical device R&D functions, causing them to become siloed and not collaborate effectively.
· Human Factors, Regulatory, Engineering, and Design teams have more in common than we think.
The medical device field is driven by risk. “Risk” means different things to different functions, and understanding these distinctions is incredibly important for enabling effective collaboration.
While there are many different aspects to doing high-quality R&D work, this article will be aimed at answering the following questions:
1) How does the FDA think about risk?
2) How do other medical device R&D functions view risk?
3) How can Human Factors help other manufacturing teams function?
How does the FDA think about risk?
I had the opportunity to attend a conference with many presenters who work for the FDA. During the conference, the FDA (fda.gov) opened by emphasizing its increased focus on risk while underscoring the complexity of defining it. These points are also evidenced by the upcoming updates to CFR 820, which focus on incorporating the most recent version of ISO 13485.
What this means for medical device manufacturers is an increased focus on accurate risk assessment in the initial medical device R&D design process, as well as creating Quality Management Systems (QMS) that can integrate real-world data and postmarket complaints. The first question to answer is, “What is Risk?”.
Searching the ISO standards risk is defined as follows:
ISO 14971: Risk – “A combination of the probability of occurrence of harm and the severity of that harm.”
In recent guidance, the FDA’s definition places less emphasis on the probability of a use error occurring and an increased emphasis on the potential harm resulting from misuse or design flaws because if something can go wrong, it will. Whether misuse/use error occurred one time or 100 times, a single instance may be enough to warrant risk evaluation if it can result in significant harm. This is why summative validation studies are required when submitting to the FDA.
Also, it is worth noting that post-market data (e.g., complaints and actual use observations) can accurately assess the risk probability. Observations of real users using the product in the field can identify new risks that weren’t foreseen in the context of design and development.
Because of the dynamic nature of real-world device use, risk must be updated throughout the medical device’s life. Even with post-market data, it is essential to recognize that many adverse events go unreported, and the same goes for off-label use, which must also be considered. The FDA expects manufacturers to assess the risk of off-label use and implement risk mitigations as needed.
The incoming update to CFR 820 means there will be an increased emphasis on the need to think through and mitigate potential risks from a user-centered perspective. This is where human factors expertise plays a crucial role in safe and effective device design.
If manufacturers stay in touch with these changes, it can prevent FDA submissions from being delayed due to inadequate risk assessment or insufficient validation. FDA audits can also harm new device sales if the risk is not adequately assessed, documented, mitigated, and affirmed through human factors validation in formative and summative studies with the intended users. This can result in flagged audits, recalls, and possible user harm.
How do different medical device R&D functions think about risk?

Different teams have their own ideas of what risk means to them. Business teams focus on the potential risk to sales and the company; marketing assesses the risk of damaging brand perception; regulatory teams focus on the risk associated with adherence to guidelines; and medical device R&D teams focus primarily on the risk to the device itself (including to device user interfaces and other components).
This can be seen in some of the tools used by each function. For example, engineers may use a dFMEA to determine the risk to the device during production. Regulatory teams use FDA guidance to determine how to assess risk across aspects of the device, like shipping or materials. Business strategy teams use earnings reports and marketing analysis to determine budget allocation. Marketing may use Voice of the Customer (VoC) or information from sales reps to determine customer perceptions of their devices.
This summation is far from complete, and there is a significant crossover in tools and priorities. Still, the point is that different teams consider different angles regarding success and risk. Every function is essential, and making a user-centered Human Factors approach a priority early in the development process can have positive cascading effects on other team functions and critical tasks.
How do Human Factors Approaches Help You and Your Team?

Human Factors professionals focus on the potential risk to users based on how they interact or may interact with the device. This includes misuse, cognitive perception, or off-label use of products. Human Factors engineers use an evaluative approach and usability engineering processes with a unique combination of qualitative and quantitative data.
Human Factors engineers then use that data to build the proper use context and assess the use-related hazards and risks associated with different use scenarios captured in a task and use-related risk analyses (URRA). A thoroughly done task analysis and URRA capture a complete picture of the final device in the context of realistic use environments with user populations by defining each step and identifying the potential for harm at each of these steps, along with residual risk.
This assessment can make identifying design shortfalls and opportunities for mitigating them much more straightforward. It can also open the door for gaining a competitive edge by addressing difficulties that are seen
as unavoidable.
For example, suppose an engineering team knows the exact steps the user will go through and the plausible errors at each step. In that case, it will enable more informed design decisions and a safer, more enjoyable final design for the user. For example, suppose an engineering team knows the steps the user will go through and the plausible errors at each step. In that case, it will enable more informed design decisions and a safer, more enjoyable final design for the user.
This testing can also aid marketing in making claims in sales material as the voice of the customer and user research process can be supercharged by Human Factors techniques such as journey mapping, in-depth user interviews, and pain points/opportunity frameworks.
A well-rounded Human Factors engineering team has experience designing for various industries and understands that the approaches and needs are nuanced for each of them. Suppose you are working in the medical device R&D field. In that case, it is crucial to have a Human Factors team that can understand the needs of and communicate clearly with engineering, design, clinical, and regulatory groups.
Having a team that stays current with new draft guidance documents from regulatory bodies you plan to submit to is also mission-critical.
Conclusion

The FDA is increasing its focus on risk to users and patients and how that risk management is executed. A well-rounded Human Factors team can help quality and regulatory teams with their submissions to pass FDA review, business and marketing teams with claims and sales, and medical device R&D teams make more informed design decisions.
While updates to regulation may seem like another hurdle, taking Human Factors into account early on in the medical device R&D process enables design changes to be made early and mitigations to be considered as needed. Employing experienced and well-informed Human Factors engineers can create opportunities for better device design while keeping up with changing FDA requirements.
If you have any questions or comments about the FDA or Human Factors in medical device development, contact us today to start the conversation.
________________________________________________________________
Ready to create opportunities for better device design while keeping up with changing FDA requirements?
________________________________________________________________
Human Factors Engineering At THRIVE
THRIVE’s Human Factors professionals have decades of experience applying Human Factors to products ranging from medical devices used by specialized healthcare professionals in clinical environments to combination products used by laypeople in the home.
If it’s your first time applying Human Factors and you need a comprehensive end-to-end Human Factors program, we’ll scope out the program and conduct the activities on your behalf, leaving you time and resources to focus elsewhere. If you’re a resource-constrained HFE professional, we’ll provide the teamwork, collaboration, and support to help you meet your goals. Or, if you want a final sanity check to ensure you’ve met the latest and greatest expectations, we’ll do that, too.
ATLANTA | CHICAGO


